Diagnosing a corneal dystrophy seems like it should be a straightforward task. You simply need to note the age of onset and clinical characteristics, ask questions about affected family members, and then run to your nearest textbook to connect the dots.
But, what if it doesn’t look exactly like the picture in the textbook? What if the inheritance pattern doesn’t make sense? Even more frustrating, what if the condition has multiple names and a variety of subtypes or clinical variants that are not agreed upon by everyone? All of a sudden, it’s not so straightforward anymore!
The term “corneal dystrophy” has been used to refer to a group of inherited corneal diseases that are typically bilateral, symmetric, slowly progressive and without relationship to environmental or systemic factors.1 But, not every disorder labeled as a “corneal dystrophy” meets the criteria set forth by the definition.
Conversely, some corneal disorders labeled as congenital defects or progressive degenerations may exhibit genetic predisposition and progression such that we should strongly consider them under the designation “corneal dystrophy.”
In reality, both the definition and the classification of corneal dystrophies have been shaped by historical clinical findings and inheritance patterns that are sometimes difficult to confirm. Today, we have a multitude of tools at our disposal to investigate the etiology of disease. Histopathological studies have improved, and current genetic analysis allows us to map the chromosomal loci and even identify the specific gene and mutation associated with each disorder. It is no surprise that these improvements in technology have made our historical classification of corneal dystrophies outdated.
Comprehensive Classification
During the World Cornea Congress V, held in Washington, D.C., in 2005, it became apparent that the then-current dystrophy classification system was inaccurate and outdated. With this in mind, the Cornea Society was approached with the idea of forming a committee to address corneal dystrophy nomenclature. In October of 2005, the first meeting of the International Committee for Classification of Corneal Dystrophies (IC3D) was held. Its goal was to develop a new classification system for corneal dystrophies, integrating up-to-date information on clinical description, pathologic examination, and genetic analysis. The result of the committee’s hard work, “The IC3D Classification of the Corneal Dystophies,” was published as a supplement to the December 2008 issue of Cornea.2
The IC3D devised a template to review our current knowledge of each corneal dystrophy in an organized manner. Each template includes the name of the dystrophy, alternative names, genetic information (e.g., the gene locus of the responsible mutation and the gene involved), onset, signs, symptoms, course, light microscopy, transmission electron microscopy, confocal microscopy and clinical photographs. The committee also categorized each corneal dystrophy based on the level of evidence available to support its existence. The IC3D does not contain epidemiological data; however, the prevalence of rare conditions, such as corneal dystrophies, is usually either unknown or simply estimated, although many are known to be more prevalent in certain populations.
The categories are as follows:
• Category 1: A well defined corneal dystrophy in which a gene has been mapped and identified and specific mutations are known.
• Category 2: A well-defined corneal dystrophy that has been mapped to one or more specific chromosomal loci, but the gene (or genes) remains to be identified.
• Category 3: A well-defined corneal dystrophy that has not yet been mapped to a chromosomal locus.
• Category 4: This category is reserved for suspected new (or previously documented) corneal dystrophies, although the evidence for such dystrophies being separate and distinct is not yet convincing.
But, there is flexibility within this categorized system. As information accumulates, all true dystrophies should eventually reach category 1. Or, some conditions that were once thought to be independent dystrophies may be later proven to be a variant of a different dystrophy. This has already occurred once in the short lifetime of this classification system. Genetic and histopathological studies of central discoid corneal dystrophy have indicated that this condition is actually Schnyder corneal dystrophy. Previously, central discoid corneal dystrophy was classified as a category 4 dystrophy, but with this new information, it has been reclassified as Schnyder corneal dystrophy.3
The IC3D classification system is organized anatomically according to which corneal layer is affected. In general, these groupings include epithelial, subepithelial, Bowman’s layer, stromal, and those effecting Descemet’s membrane and the endothelium. The system also groups dystrophies together if they have a common genetic relationship. For example, there have been over 30 mutations in the transforming growth factor beta-induced gene (TGFBI) that have been found to cause types of lattice corneal dystrophy and granular corneal dystrophy.4 So, these dystrophies are grouped together. (For the purposes of this article, we will focus on of the aspects of these disorders that you can observe in a normal exam room. But for those interested, a brief summary of the genetics can be found in Table 1, below.)
Table 1: A Brief Summary of the Genetics of Corneal Dystrophies
| Dystrophy
|
Mode of Inheritance
|
Gene Locus | Gene
|
IC3D Category |
| Epithelial and subepithelial dystrophies
| ||||
| Epithelial basement membrane dystrophy (EBMD)
|
Mostly sporadic
|
5q31
|
TGFBI in minority of cases
|
1 in minority of cases
|
| Epithelial recurrent erosion dystrophy (ERED)
|
AD | Unknown
|
Unknown
|
4,3 (Smolandiensis)
|
| Subepithelial mucinous corneal dystrophy (SMCD)
|
AD
|
Unknown | Unknown
|
4
|
| Meesman corneal dystrophy (MECD)
|
AD
|
12q12, 17q12 (Stocker-Holt)
|
KRT3, KRT12 (Stocker-Holt)
|
1 including Stocker-Holt
|
| Lisch epithelial corneal dystrophy
|
XD
|
Xp 22.3
|
Unknown
|
2
|
| Gelatinous drop-like corneal dystrophy
|
AR
|
Ip32
|
TACSTD2
|
1
|
| Bowman's layer dystrophies
| ||||
| Reis-Bucklers corneal dystrophy
|
AD
|
5q31
|
TGFBI
|
1
|
| Thiel-Benke corneal dystrophy
|
AD
|
10q24
|
Unknown
|
2
|
| Grayson-Wilbrandt Corneal Dystrophy
|
AD
|
Unknown
|
Unknown
|
4
|
| Stromal dystrophies
| ||||
| Lattice corneal dystrophy, type 1 and variants
|
AD
|
5q31
|
TGFBI | 1
|
| Granular corneal dystrophy, type 1
|
AD
|
5q31
|
TGFBI | 1
|
| Macular corneal dystrophy
|
AR
|
16Q22 | CHST6
|
1 |
| Schnyder corneal dystrophy
|
AD
|
1p36 | UBIAD1
|
1 |
| Congenital stromal corneal dystrophy
|
AD
|
12q31.33 | DCN
|
1 |
| Fleck corneal dystrophy
|
AD
|
2q35
|
PIP5K3
|
1 |
| Posterior amorphous corneal dystrophy
|
AD
|
Unknown
|
Unknown
|
3
|
| Central cloudy dystrophy of Francois
|
Unknown
|
Unknown
|
Unknown
|
4
|
| Pre-Descemet corneal dystrophy
|
Unknown
|
Unknown
|
Unknown
|
4
|
| Descemet's membrane and endothelial dystrophies
| ||||
| Fuch's endothelial corneal dystrophy, late onset
|
Unknown, some AD
|
13pTel-13q12.13, 15q, 18q21.2-q21.23
|
Unknown
|
2
|
| Fuch's endothelial corneal dystrophy, early onset
|
AD
|
Ip34.3
|
COL8A2
|
1 |
| Posterior polymorphous corneal dystrophy 1
|
AD | 20p11.12-q11.2 | Unknown
|
2 |
| Posterior polymorphous corneal dystrophy 2 | AD | Ip34.3-p32.3 | COL8A2 | 1 |
| Posterior polymorphous corneal dystrophy 3 | AD | 10p11.12 | ZEB1
|
1 |
| Congenital hereditary endotholial dystrophy 1
|
AD
|
20p11.12-q11.2 | Unknown | 2 |
| X-linked endothelial corneal dystrophy
|
XS
|
Xq25
|
Unknown
|
2 |
Epithelial and Subepithelial Dystrophies
Figure 1 shows a fairly typical example of the clinical appearance of epithelial basement membrane dystrophy (EBMD). Although the presentation is somewhat variable, common findings include dot-like epithelial opacities, whorl-like fingerprint lines, and circumscribed grey map-like patterns. Particular forms of EBMD are category 1—namely, the few recorded cases that show an autosomal-dominant inheritance pattern and involve a mutation in the TGFBI gene. The vast majority of EBMD cases, however, are thought to be sporadic, and they may represent a degenerative condition rather than a true dystrophy. Recurrent corneal erosion (RCE) is a common complication of EBMD. If medical management of RCE fails, phototherapeutic keratectomy (PTK) or epithelial debridement and polishing of Bowman’s layer using a diamond burr have been proven effective treatments.5-7
Epithelial recurrent erosion dystrophy (ERED) is characterized by RCE that occurs spontaneously or after minimal trauma. The attacks have been documented in patients as young as eight months, but usually appear around four to six years of age. In most cases, the attacks decline in frequency and intensity by 50 years of age. But in the Smolandiensis variant, half of patients develop progressive central subepithelial opacities as early as age seven. In these cases, corneal erosions are treated medically, and a corneal graft is needed in about one-fourth of patients.8-11
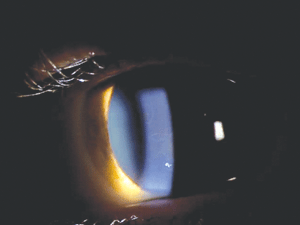
Figure 1: Map-like opacities in EBMD.
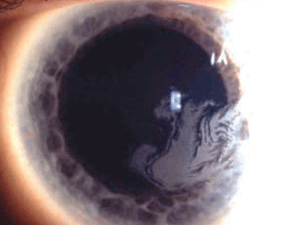
Figure 2: Whorl-like grey opacity in Lisch Epithelial Corneal
Dystrophy. (Reproduced from IC3D Classification of the Corneal
Dystrophies with permission from The Cornea Society).
Lisch epithelial corneal dystrophy (LECD) usually presents in childhood with gray opacities and microcysts in the epithelium arranged in feathery, radial, flame-shaped, band-shaped, or whorl-like patterns (figure 2). It is the only known corneal epithelial dystrophy with X-chromosomal dominant inheritance. The opacities can progress to produce vision loss. Debridement of the corneal epithelium does not prevent the opacities from recurring, but rigid contact lenses have been shown to lessen the number of opacities in some patients.12-14
Subepithelial mucinous corneal dystrophy presents with subepithelial opacities and haze that is most dense centrally. Recurrent corneal erosion can also occur, though it usually decreases in frequency during adolescence. Only one case, of a single family, is reported in the literature.15
Meesmann corneal dystrophy (MCED) is a category 1 dystrophy characterized by numerous tiny gray epithelial vesicles that extend to the limbus but are often denser in the interpalpebral zone. Signs of this autosomal dominant dystrophy usually appear in infancy or early childhood. MCED progresses slowly and is often asymptomatic until middle age.
Symptoms often include mild ocular irritation and light sensitivity; some patients experience blurred vision due to corneal surface irregularity and scarring from erupted vesicles. The Stocker-Holt variant of MCED presents with more severe signs and symptoms, as well as fine, punctate epithelial opacities that stain with fluorescein and infrequent linear opacities in whorl-like patterns. Treatment depends on severity, and can include lubricants, therapeutic contacts, epithelial debridement, PTK and lamellar keratoplasty (LK).16-20
Gelatinous drop-like corneal dystrophy is an autosomal recessive category 1 corneal dystrophy found mostly in Japanese patients. It presents with subepithelial, milky white, gelatinous, mulberry-shaped nodules in the first or second decade of life. Initially, these lesions can resemble band-shaped keratopathy, and they often stain late with fluorescein. Progressive stromal opacification, or the development of large nodular kumquat-like lesions, often causes significant decreased vision. Contact lenses can help manage corneal surface irregularities. Superficial keratectomy (SK), LK, or PK have all been used in treatment, but the dystrophy often recurs within a few years. Limbal stem cell transplantation, along with PK or LK, has shown some success in preventing recurrence.21-23
Bowman’s Layer Dystrophies
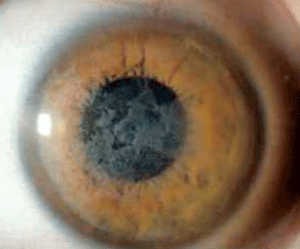
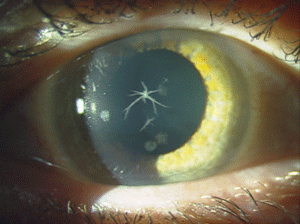
Figure 3: Coarse
geographic opacities in Reis-Bücklers corneal dystrophy. (Reproduced
from IC3D Classification of the Corneal Dystrophies with permission
from The Cornea Society).
Reis-Bücklers corneal dystrophy (RBCD) and Thiel-Benhke corneal dystrophy (TBCD) are both category 1 dystrophies with autosomal dominant inheritance and similar clinical findings (figure 3). They both present in childhood with reticular superficial corneal opacities (described as “honeycomb-like” in TBCD). These opacities, along with superficial corneal irregularity and RCE, can cause slow, progressive deterioration of vision. But, the course of RBCD is often more aggressive than that of TBCD. Due to their clinical similarities, electron microscopy is needed to differentiate them. Therapeutic contact lenses, medical management of RCE, SK, PK, and PTK have all been used in the treatment of RBCD, but recurrence rates are high. Mytomycin C may aid in preventing recurrence when used in conjunction with PTK in RBCD.24-30
Grayson-Wilbrandt Corneal Dystrophy (GWCD) is described as having variable patterns of opacification that extend anteriorly into the epithelium, as well as refractile bodies in the stroma. It is a good example of a category 4 dystrophy because there has only been one publication that describes only a single family with this condition, and the limited information we have on this condition does not rule out the possibility that it could be a variant of a better-described dystrophy, such as RBCD.31
Stromal Dystrophies
The IC3D describes two distinct types of lattice corneal dystrophy (LCD), although both have variant forms and presentations. The first, lattice corneal dystrophy Type 1 (LCD1) is a “true” corneal dystrophy, caused by numerous different mutations in the TBFBI gene. Lattice corneal dystrophy gelsolin type 2 (LCD2) is actually the result of a systemic familial amyloidosis with numerous systemic findings, and therefore is not a true corneal dystrophy per the definition above. LCD1 is a more common corneal dystrophy found in a variety of Asian and European populations, where LCD2 is more rare and found mostly in people of Finnish decent. The classic refractile lines or dots are seen in both disorders, but in LCD1, they generally are more prominent centrally and leave the peripheral 11mm of cornea spared, whereas in LCD2, they are more peripheral and less numerous. Ocular signs and symptoms in LCD1 are generally notable within the first decade of life, while LCD2 generally becomes apparent after the age of 20. In LCD1, recurrent erosions are common, and visual deterioration often requires corneal transplantation after the fourth decade of life. Recurrence generally occurs in grafts two to 14 years later. Corneal transplantation is rarely indicated in LCD2; neurotrophic persistent epithelial defects after PK have been described. LK and PTK can also be used in the treatment of LCD1, but as with PK, the dystrophy eventually recurs.32-39
|
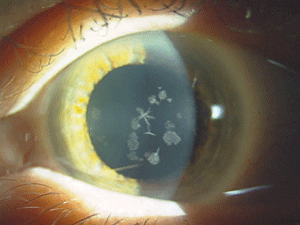
|
| Figure 4: Star-shaped
and breadcrumb-like opacities in the eyes of a patient with Granular
Corneal Dystrophy Type 2. (Photos courtesy of Jason Jedlicka, O.D.)
| |
Recently, a paper published in Cornea discussed a 65-year-old man with the what seemed to be a classic case of LCD. However, all known mutations of the TGFBI gene were ruled out, and a myeloproliferative disorder, monoclonal gammopathy, was found to be the cause. Whether or not the IC3D will consider this as another subtype of LCD remains to be seen.40
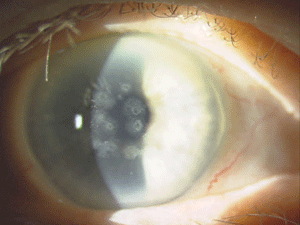
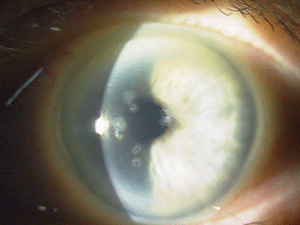
Figure 5: These are photos of the mother of the patient in Figure
4. Note the slightly different presentation with primarily disc-shaped
and breadcrumb-like opacities. (Photos courtesy of Jason Jedlicka,
O.D.)
Granular corneal dystrophy (GCD) is also separated into two distinct subtypes, both with category 1 levels of evidence. Both autosomal-dominant subtypes are caused by mutations in the TGFBI gene. Granular corneal dystrophy type I (GCD1) presents within the first decade of life and develops into the typical stromal, white, sharply demarcated lesions that resemble bread crumbs or snowflakes. Granular corneal dystrophy type 2 (GCD2) often has fewer deposits than GCD1, but it may present with deep lattice lines in combination with the more superficial breadcrumb-like deposits seen in GCD1 (figures 4 and 5). GCD2 is usually diagnosed in the teens or early adulthood and often progresses more slowly than GCD1. Although not all patients with GCD develop vision loss that necessitates surgery, PK, LK and PTK have all been used. However, most forms of laser and/or refractive surgery have been found to merely exacerbate GCD2.41-45
Macular corneal dystrophy (MCD) is a category 1 autosomal recessive dystrophy that presents in childhood with diffuse stromal haze that extends to the limbus (figure 6). Over time, central, whitish, elevated opacities develop and coalesce, leaving no clear interval between them.
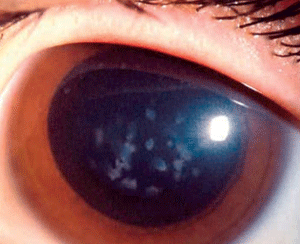
Figure 6: Stromal
opacities in the central cornea in Macular Corneal Dystrophy.
(Reproduced from IC3D Classification of the Corneal Dystrophies with
permission from The Cornea Society)
In advanced stages of the disease, nearly all layers of the cornea are affected. Guttata-like lesions form in Descemet’s membrane, and endothelial dysfunction leads to stromal thickening. RCE and corneal surface irregularity can occur. Vision is severely affected between 10 years to 30 years of age.
Gas-permeable lenses can help in cases of irregular astigmatism, but PTK and eventually PK are often needed to restore corneal clarity.46-48
Schnyder corneal dystrophy is a category 1 autosomal-dominant dystrophy that is usually apparent by the second or third decade (figure 7). Corneal haze and/or subepithelial crystals are noted in patients 23 years of age and younger. However, only 50% of patients develop crystals, and they may occur later in the disease. Systemic hypercholesterolemia is common, and arcus lipoides is usually noted between the ages of 23 and 38. Slowly progressive corneal opacification in a central discoid pattern often occurs. Surprisingly, scotopic vision is often quite good, but reduction in photopic vision necessitates corneal transplantation for the majority of patients over 50 years of age.49-53
Congenital stromal corneal dystrophy (CSCD) is another autosomal-dominant category 1 corneal dystrophy. As the name suggests, it presents at birth with diffuse clouding of the cornea and whitish flake-like stromal deposits. It is extremely rare; only four families have documented cases. This dystrophy progresses slowly or remains stationary and can produce moderate to severe vision loss.54-56
Fleck corneal dystrophy (FCD) is a category 1 autosomal-dominant dystrophy that is non-progressive and asymptomatic. It presents with small gray-white dandruff-like flecks or translucent discoid opacities scattered sparsely throughout any level of the stroma.57-59
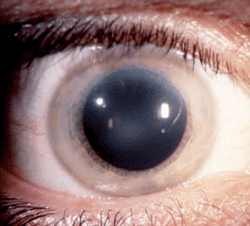
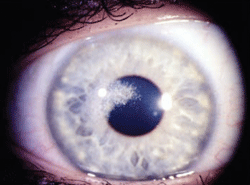
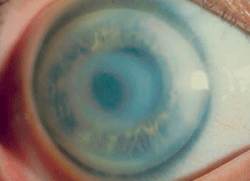
Figure 7: Schnyder
Corneal Dystrophy (SCD). A. Early opacity. B. Early opacity with
crystals. C Central ring shaped opacity with prominent peripheral arcus
lipoids. (Reproduced from IC3D Classification of the Corneal
Dystrophies with permission from The Cornea Society)
Posterior amorphous corneal dystrophy (PACD) is an autosomal-dominant category 3 dystrophy that presents with gray-white, sheet-like opacities that can involve any layer of the stroma. PACD has been observed as early as 16 weeks of age. Corneal flattening or thinning and endothelial opacities may also be present. A prominent Schwalbe’s line, as well as iris and angle findings, may also be present. The possible congenital onset and lack of progression make it unclear whether this is actually a developmental disorder or a corneal dystrophy.60-62
Central cloudy dystrophy of Francois (CCDF) is a category 4 dystrophy that closely resembles posterior crocodile shagreen. Many cases in the literature do not provide verification that this is a familial disorder, so it is possible that CCDF is actually the corneal degeneration posterior crocodile shagreen. Usually, this condition is asymptomatic, and one case report in the literature shows that LASIK has been used to treat refractive error in a patient with CCDF without complication.63-65
Pre-Descemet’s corneal dystrophy (PDCD) is another ill-defined category 4 corneal dystrophy with different subtypes (figure 8). No definite familial inheritance is noted, and vision is usually minimally affected. The corneal findings include focal, fine, gray opacities in the deep stroma immediately anterior to Descemet’s membrane in a variety of shapes. But, more uniformly distributed polychromatic opacities are noted in one subtype. Similar opacities have been noted in association with other ocular and systemic diseases, especially ichthyosis and carriers of X-linked ichthyosis. For this reason, it is unclear whether PDCD is a dystrophy or degeneration.66-68
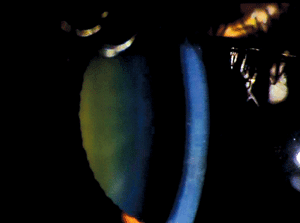
Figure 8: Fine gray opacities just anterior to Descemet membrane consistent with Pre-Descemet Corneal Dystrophy.
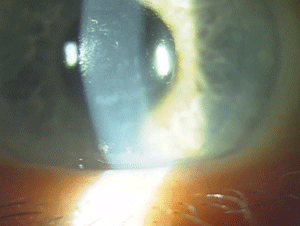
Figure 9: Endothelial changes in Posterior Polymorphous Corneal Dystrophy. (Photo courtesy of Jason Jedlicka, O.D.)
Fuchs’ endothelial corneal dystrophy (FECD) presents with corneal guttata, stromal edema and oftentimes, endothelial pigment. This dystrophy is familiar to most of us, but the genetics for all types of Fuchs’ are not completely worked out. Autosomal dominant inheritance has been reported, but many patients have no known inheritance pattern. The IC3D has designated different varieties of Fuchs’ as category 1, 2, and 3. Category 1: early onset Fuchs’, which is usually notable by the first decade and typically presents with smaller guttata than late onset Fuchs’. In most cases of Fuchs’, onset is in the fourth decade or later. Another novel genetic locus has been linked to a mild form of late-onset Fuchs’ since the IC3D classification was published, and new publications on both the treatment and genetics of Fuchs’ make it an exciting subject in eye care right now.69-73
The IC3D recognizes three types of posterior polymorphous corneal dystrophy (PPCD) due to the variability in their genetics. Most cases demonstrate autosomal-dominant inheritance, but isolated unilateral cases show no heredity, according to the IC3D. Deep corneal lesions with nodular, vesicular or blister-like shapes are present, as well as linear lesions that resemble railroad tracks (figure 9). Approximately 25% of patients show iridocorneal adhesions, and 15% have elevated intraocular pressure. Many times PPCD is fairly stationary; however, if patients do progress, treatment options are similar to those for FECD.74-76
Congenital hereditary endothelial dystrophy type 1 (CHED 1) is an autosomal-dominant category 2 dystrophy that presents congenitally or within the first two years of life with clouding and thickening of the cornea. Progression of corneal clouding generally occurs over one to 10 years. However, there is a subset of patients who only have asymptomatic endothelial changes in a moon crater-like appearance.74,75,77
In contrast, congenital hereditary endothelial dystrophy type 2 (CHED2) is an autosomal-recessive category 1 dystrophy that is truly congenital. CHED2 presents with findings similar to those of CHED1, but it’s more common and often, more severe. PK may be indicated in either form of CHED.78,79 X-linked endothelial corneal dystrophy (XECD) is a X-chromosomal dominant congenital dystrophy that presents findings similar to those of CHED1 and CHED2 in males. Males may also have associated nystagmus and band keratopathy. But in females, moon crater-like endothelial changes are often the only finding. Males often have blurred vision, and the condition is progressive. Females are usually asymptomatic, and the disease is nonprogressive. PK has been performed on males with XECD.80
A Basis for Further Understanding
I hope this brief description of the corneal dystrophies has served as an introduction and an invitation to read the more comprehensive work: IC3D Classification of the Corneal Dystrophies. The IC3D has formulated a concise, well-organized classification system that helps to clarify much of the confusion surrounding corneal dystrophies. It has proven itself to be an extremely useful clinical reference and will act as a repository for new information in the field.
Dr. Veire practices optometry with a large medical group in St. Paul, Minn. He has special interests in contact lenses and ocular surface disease. He can be contacted at [email protected].
1. American Academy of Ophthalmology. External disease and cornea. In: Sutphin J, ed. Basic and Clinical Sciences Course 2007-2008. San Francisco, CA: American Academy of Ophthalmology; 2007: 305-329.
2. Weiss J, Møller H, Lisch W, et al. The IC3D classification of the corneal dystrophies. Cornea. 2008;27:S1-S42.
3. Ridgway A, Akhtar S, Munier F, et al. Ultrastructural and molecular analysis of Bowman’s layer corneal dystrophies: an epithelial origin? Invest Ophthalmol Vis Sci. 2000; 41:3248-92.
4. Kannabiran C, Klintworth G. TGFBI Gene mutations in corneal dystrophies. Human Mutation. 2006;27:615-625.
5. Kanski J. Clinical Ophthalmology: a systemic approach, 6th edition. Philadelphia PA: Heinemann: Butterworth; 2007: 291-302.
6. Boutboul S, Black G, Moore J, et al. A subset of patients with epithelial basement membrane corneal dystrophy have mutations in TGFBI/BIGH3. Human Mutation 2006; 6:553-57.
7. Sridhar M, Rapuano C, Cosar C, et al. Phototherapeutic keratectomy versus diamond burr polishing of Bowman’s membrane in the treatment of recurrent corneal erosions associated with anterior basement membrane dystrophy. Ophthalmology. 2002; 109:674-9.
8. Hammer B, Bjorck E, Langerstedt K, et. al. A new corneal disease with recurrent erosive episodes and autosomal dominant inheritance. Acta Ophthalmol. 2008; 86:758-63.
9. Franceschetti A. Hereditäre rezidivierende Erosion der Hornhaut. Augenheilk. 1928; 66:309-16.
10. Valle O. Hereditary recurring corneal erosions: a family study with special reference to Fuchs’ dystrophy. Acta Ophthalmol. 1967; 45:829-36.
11. Wales HJ: A family history of corneal erosions. Trans Ophthalmol Soc NZ. 1956; 8:77-78.
12. Lisch W, Steuhl K, Lisch C, et al. A new, band-shaped and whorled microcystic dystrophy of the corneal epithelium. Am J Ophthalmol. 1992; 114:35-44.
13. Alvarez-Fischer M, Alvarez de Toledo J, Barraquer R. Lisch corneal dystrophy. Cornea. 2005;24:494-95.
14. Butros S, Lang G, Alvarez de Toledo J, et al. The different opacity patterns of Lisch corneal dystrophy. Klin Monbl Augenheilkd. 2006 Oct; 223:837-40.
15. Feder R, Jay M, Yue B, et al. Subepithelial mucinous corneal dystrophy. Clinical and pathological correlations. Arch Ophthalmol. 1993; 111:1106-1114.
16. Meesmann A. Über eine bisher nicht beschriebebe dominant vererbte Dystrophia epithelialis corneae. Ber Zusammenkunft Dtsch Ophthalmol Ges. 1938; 52:154-58.
17. Fine B, Yanoff M, Pitts E, et al. Meesmann’s epithelial dystrophy of the cornea. Am J Ophthalmol. 1977; 83:633-42.
18. Burns R. Meesmann’s corneal dystrophy. Trans Am Ophthalmol Soc. 1968;66:530-635.
19. Stocker F, Holt L. A rare form of hereditary epithelial dystrophy of the cornea: a genetic, clinical and pathological study. Trans Am Ophthalmol Soc. 1954;52133-44.
20. Jalbert I, Stapleton F. Management of Symptomatic Meesmann Dystrophy. Optom Vis Sci. 2009. Sept 7.
21. Weber F, Babel J. Gelatinous drop-like dystrophy. A form of primary corneal amyloidosis. Arch Ophthalmol 1980; 98:144-48.
22. Ide T, Nischida K, Maeda N, et al. A spectrum of clinical manifestations of gelatinous drop-like corneal dystrophy in Japan. Am J Ophthalmol. 2004; 137:1081-84.
23. Shimazaki J, Shimmura S, Tsubota K. Limbal stem cell transplantation for the treatment of supepithelial amyloidosis of the cornea (gelatinous drop-like dystrophy). Cornea. 2002; 21:177-80.
24. Bücklers M. Über eine weitere familiäre Hornhaut-dystrophie (Reis). Klin Manotsbl Augenkeilkd. 1949;114:386-97.
25. Reis W. familiäre, fleckige Hornhautentartung. Disch Med Wocheschr. 1917; 43:575.
26. Küchle M, Green W, Völcker H, et al. Reevaluation of corneal dystrophies of Bowman’s layer and the anterior stroma (Reis-Bückler’s and Theil-Behnke types: a light and electron microscope study of eight corneas and a review of the literature. Cornea. 1995; 14:333-54.
27. Theil H. Behnke H. Eine bisher unbekannte subepitheliale hereditare Hornhautdystrophie. Klin Monatsble Augenheilkd. 1967; 150:862-74.
28. Kobayashi A, Sugiyama K. In vivo laser confocal microscopy findings for Bowman’s layer dystrophies (Thiel-Behnke and Reis- Bücklers corneal dystrophies). Ophthalmology. 2007; 114:69-75.
29. Orndahl M, Fagerholm P. Treatment of corneal dystrophies with phototheraputic keratectomy. J Refract Surg. 1998;14: 129-35.
30. Shwartz M, Tayler H. Surgical management of Reis- Bücklers’ corneal dystrophy. Cornea. 1985; 4:100-7.
31. Grayson M, Wilbrandt H. Dystrophy of the anterior limiting membrane of the cornea (Reis-Bückler’s type). Am J Opthalmol. 1966; 61:345-49.
32. Biber H. Ueber einige seltene Hornhautkrankungen:die oberflächliche gittrige Keratitis (inaugural dissertation). Zurich; 1890.
33. Klintworth, GK: Lattice corneal dystrophy. An inherited variety of amyloidosis restricted to the cornea. Am J Pathol. 1967; 50:371-99.
34. Stock E, Feder R, O’Grady R, et al. Lattice corneal dystrophy type IIIA. Clinical and histopathologic correlations. Arch Optholmol. 1991; 109:354-58.
35. Vajpayee R, Tyagi J, Sharma N, et al. Deep anterior lamellar keratoplasty by big-bubble technique for treatment corneal stromal opacities. Am J Ophthalmol. 2007; 143:954-57.
36. Meretoja J. Familial systemic paramyloidosis with lattice dystrophy of the cornea, progressive cranial neuropathy, skin changes and various internal symptoms. A previously unrecognized heritable syndrome. Ann Clin Res. 1969; 1:314-24.
37. Kivelä T, Tarkkanen A, Frangione B, et al. Ocular amyloid deposition in familial amyloidosis, Finnish: an analysis of native and variant gelsolin in Meretoja’s syndrome. Invest Ophthalmol Vis Sci. 1994; 35:3759-69.
38. Kawamoto K, Morishige N, Yamada N, et al. Delayed corneal epithelial wound healing after penetrating keratoplasty in individuals with lattice corneal dystrophy. Am J Ophthalmol. 2006;142:173-74.
39. Das S,Langenbucher A, Seitz B. Excimer laser phototherapeutic keratectomy for granular and lattice corneal dystrophy: a comparative study. J Refract Surg. 2005; 21:727-31.
40. Kamal K, Rayner S, Chen M, et al. Classic lattice corneal dystrophy associated with monoclonal gammopathy after exclusion of a TGFI mutation. Cornea. 2009; 28:97-98.
41. Groenouw A. Knötchenförmige Hornhauttrübungen (nodili corneae). Arch Augenheilkd. 1890; 21:281-89.
42. Møller H. Granular corneal dystrophy Groenouw type I. Clinical and genetic aspects. Acta Opthalmol. 1991;69 (suppl 198);1-40.
43. Holland E, Daya S, Stone E, et al. Avellino corneal dystrophy. Clinical manifestations and natural history. Ophthalmology. 1992; 99:1564-68.
44. Jun R, Tchah H, Kim T, et al. Avellino cornal dystrophy after LASIK. Ophthalmol. 2004; 111:463-68.
45. Lee J, Sulting R, Lee D, et al. Exacerbation of granular corneal dystrophy type II (Avellino corneal dystrophy) after LASEK. J Refract Surg. 2008; 24:39-45.
46. Klintworth, G, Vogel F. Macular corneal dystrophy: an inherited acid mucopolysaccharide storage disease of the corneal fibroblast. Am J Pathol. 1964; 45:565-86.
47. Jonasson F, Oshima E, Thonar E, et al. Macular corneal dystrophy in Iceland. A clinical, genealogic, and immunohistochemical study of 28 patients. Ophthalmology. 1996; 103:111-17.
48. Gruenauer-Kloevekorn C, Braeutigam S, Heinritz W, et al. Macular corneal dystrophy: mutational spectrum in German patients, novel mutations and therapeutic options. Graefes Arch Clin Exp Ophthalmol. 2008: 246:1441-7
49. Schnyder W. Mitteilung über einen neuen Typus von familiärer Hornhauterkrankung. Schweiz Med Wschr. 1929; 10: 559-517.617.
50. Lisch W, Weidle G, Lisch C et al. Schnyder’s dystrophy. Progression and metabolism. Ophthalmic Pediatr Genet. 1986; 7: 45-56.
51. Weiss J, Rodrigues M, Kruth H et al. Panstromal Schyder’s corneal dystrophy. Ultrastructure and histochemical studies. Ophthalmology. 1992;99:1072-81.
52. Weiss J. Schnyder’s dystrophy of the cornea. A Swede-Finn connection. Cornea. 1992; 11:83-101.
53. Weiss J, Visual morbidity in thirty-four families with Schnyder’s corneal dystrophy. Trans Am Soc Ophthamol Vis Sci. 2007; 105:1-33.
54. Witschel H, Fine B, Grützner P, et al. Congenital hereditary stromal dystrophy of the cornea. Arch Ophthalmol. 1978; 96: 1043-51.
55. Van Ginderdeuren R, De Vos R, Casteels I, et al. Report of a new family with dominant congenital hereditary stromal dystrophy of the cornea. Cornea. 2002;21:118-20.
56. Bredrup C, Knappsckog P, Majewski J, et al. Congenital stromal dystrophy of the cornea caused by a mutation in the decorin gene. Invest Ophthalmol Vis Sci. 2005; 46:420-26.
57. Purcell J, Krachmer J, Weingeist T. Fleck corneal dystrophy. Arch Ophthalmol. 1977; 95:440-44.
58. Kiskaddon B, Campbell R, Waller R. Fleck dystrophy of the cornea: case report. Ann Ophthalmol. 1980; 12:700-04.
59. Assi A, Ebenezer N, Ficker L. Corneal fleck dystrophy in an English family. Br J Ophthalmol. 1999; 83:1407-8.
60. Carpel EF, Sigelman R, Doughman D. Posterior amorphous corneal dystrophy. Am J Ophthalmol. 1977; 83:629-32.
61. Dunn S, Krachmer J, Ching S. New findings in posterior amorphous corneal dystrophy. Arch Ophth. 1984; 102:236-39.
62. Johnson AT, Folberg R, Vrabec M, et al. The pathology of posterior amorphous corneal dystrophy. Ophthalmology. 1990; 97:104-09.
63. François C, Scott, I, Green W, et al. Une nouvelle dystophie hérédo-familiale de la cornée. J. Genet Hum. 1956; 5:189-96.
64. Meyer J, Quantock A, Thonar E, et al. Characterization of a central corneal cloudiness sharing features of posterior crocodile shagreen and central cloudy dystrophy of François. Cornea. 1996; 15:347-54.
65. Zaidi A, Mcleod S. Laser in situ keratomileusis in a patient with presumed central cloudy corneal dystrophy of François. Am J Ophthalmol. 2005; 139:37677.
66. Curran R, Kenyon, K Green W. Pre-descemet’s membrane corneal dystrophy. Am J Ophthalmol. 1974; 77:7110-16.
67. Ferandez-Sasso D, Acosta J, Malbran E. Punctiform and polychromatic pre-Descemet’s dominant corneal dystrophy. Br J Ophthalmol. 1979; 63:336-38.
68. Kempster R, Hirst L, dela Cruz Z, et al. Clinicopathologic study of the cornea in X-linked ichthyosis. Arch Opthalmol. 1997; 115:409-15.
69. Fuchs E. Dystrophia epithelialis cornea. Albrecht von Graefes Arch Clin Exp Ophthalmol. 1910; 76:478-508.
70. Krachmer J, Purcell J, Joung C, et al. Corneal endothelial dystrophy. A study of 64 families. Arch Ophthalmol. 1978; 96: 2036-39.
71. Sundin O, Jun A, Broman K, et al. Linkage of late-onset Fuchs corneal dystrophy to a novel locus at 13ptel-13q12.13. Invest Ophthalmol Vis Sci. 2006; 47:140-45.
72. Sudin O, Broman K, Chang H, et al. A common locus for late-onset Fuchs corneal dystrophy maps to 18q21.2-q21.32. Invest Ophthalmol Vis Sci. 2006; 47:3912-20.
73. Riazuddin S, Eghrari A, Al-Saif A, et al. Linkage of mild late-onset phenotype of Fuchs corneal dystrophy to a novel locus at 5q33.1-q35.2. Invest Ophthalmol Vis Sci. 2009; 50:5667-71.
74. Cibis G, Krachmer J, Phelps C, et al. The clinical spectrum of posterior polymorphous dystrophy. Arch Ophthalmol. 1977; 95:1529-37.
75. McCarney A, Kirkness C. Comparison between posterior polymorphous dystrophy and congenital hereditary endothelial dystrophy of the cornea. Eye. 1988; 2:63-70.
76. Aldave A, Yellore V, Pricipe A, et al. Candidate gene screening for posterior polymorphous dystrophy. Cornea 2005; 24:151-55.
77. Pearce W, Tripathi R, Morgan G. Congenital endothelial corneal dystrophy. Clinical, Pathological, and genetic study. Br J Ophthalmol. 1969; 53;577-91.
78. Maumenee A. Congenital hereditary corneal dystrophy. Am J Opthalmol. 1960; 50:1114-24.
79. Kirkness C, McCartney A, Rice N, et al. Congenital hereditary corneal edema of Maumenee: its clinical fetures, management, and pathology. Br J Opthalmol. 1987; 71:130-44.
80. Schid E, Lisch W, Philipp W, et al. A new, X-linked endothelial corneal dystrophy. Am J Opthalmol. 2006; 141:478-87.

