While the answer to the first question is beyond the scope of this article, several methods exist for organizing congenital corneal anomalies. The acronym STUMPED can be a successful tool to help clinicians recall the various diagnoses:1,2
S — Sclerocornea and other anomalies of Size
T — Tears and Trauma
U — Ulcers
M — Metabolic
P — Peters’ anomaly and other mesodermal dysgenesis syndromes
E — Edema
D — Dermoids and Dystrophies
These corneal anomalies are almost always diagnosed on clinical findings. Additionally, obtaining adjunct testing on pediatric patients can be quite challenging. A firm grasp on the clinical presentations of many of these corneal irregularities is key to a proper diagnosis.
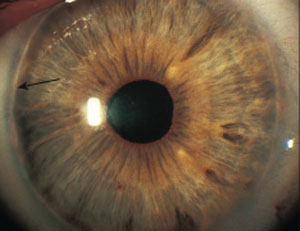 |
| An irregular white line just concentric with and anterior to the limbus (black arrow) presents with posterior embryotoxon. |
Clinically, sclerocornea is a rare, bilateral clouding of the cornea. The white appearance is consistent with that of the sclera and may be limited to the periphery or spread centrally toward the visual axis. The central cornea tends to be clearer than the limbus, which is nearly indistinguishable from sclera.1,3
Megalocornea and microcornea also represent rare, often bilateral corneal abnormalities. Megalocornea is diagnosed by a corneal diameter greater than 13mm. It is most often seen in males, as it is linked to the X-chromosome. While the cornea is histologically normal in these cases, clinicians should take care to distinguish the condition from buphthalmos, in which the cornea bulges forward secondary to high intraocular pressure (IOP).
Microcornea, conversely, presents with a corneal diameter of less than 10mm. While the cornea is also normal in this case, the condition is often accompanied by cornea plana and a shallow anterior chamber. Both anomalies require long-term monitoring for glaucoma due to concurrent angle anomalies.4,5
Tears in Descemet’s Membrane or Trauma
Forceps delivery and birth trauma can each cause breaks in Descemet’s membrane. Although forceps delivery is less common today than in the past, clinicians may still see its ocular effects in older patients. These patients will typically present with a unilateral corneal opacity with overlying corneal edema (stromal and epithelial).1,3 Careful attention to patient history and other signs of trauma will aid in confirming the diagnosis. Edema will ultimately leave pathognomonic vertical or oblique scars in Descemet’s membrane. These scars should be differentiated from Haab’s striae, which are horizontal Descemet’s folds seen in congenital glaucoma.6,7
 |
| In Reiger’s anomaly, defects such as iris strands can span the angle and attach to the posterior embryotoxon. |
Corneal ulcers should be ruled out in any patient presenting with a corneal opacification.1 Pediatric microbial keratitis is rare but potentially devastating, as pediatric eyes mount a severe inflammatory response. These ulcers are most often caused by herpetic or bacterial infections, and they warrant immediate referral for treatment. While pediatric treatment is generally the same as adult treatment, it requires extra care to ensure nontoxicity due to blood absorption.8
Metabolic Conditions
Children with corneal deposits may present with several congenital metabolic disorders:
Cystinosis. This rare autosomal recessive disease results in widespread deposition of cysteine crystals in the cornea, conjunctiva, iris, lens and retina. Aside from potentially fatal renal failure, corneal crystals are often the first extra-renal findings.4,5,9
Mucopolysaccharidosis. Caused by a missing enzyme, this lysosomal disorder breaks down glycosaminoglycan.1-3 Patients present with bilateral corneal opacities, which can be diffuse. Additional findings of glaucoma, retinal abnormalities and optic nerve swelling, all of which can be confirmed with laboratory studies or conjunctival biopsies, may aid in diagnosis.1
Fabry’s disease. This lysosomal storage disorder results in pathognomonic vortex keratopathy and wedge-shaped cataracts. Corneal findings can be differentiated from medication-induced deposits of similar appearance based on pharmaceutical history. Conjunctival and retinal vascular changes, third nerve palsy and nystagmus may support the diagnosis, and classic skin findings such as angiokeratoma corporis diffusum can confirm diagnosis.4,6
Tyrosinemia type 2. High plasma tyrosine levels that cause a recalcitrant pseudodendritic keratitis characterize this condition. Palmar and plantar hyperketatotic lesions are common and can aid in diagnosis.4,6
Wilson’s disease. This is a caeruloplasmin deficiency that leads to widespread copper deposition throughout the body. Clinical signs include Kayser-Fleischer rings, which are darker versions of arcus or sunflower cataracts or both. These patients also suffer from liver disease, basal ganglia dysfunction and psychiatric disturbance.4,6
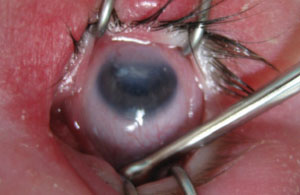 |
| Corneal opacity and iridocorneal adhesions are common presentations with Peters’ anomaly. |
Peters’ anomaly is the final stage in a group of bilateral hereditary disorders known as the mesodermal dysgenesis syndromes, which also includes posterior embryotoxon and Axenfeld-Rieger syndrome. All three entities arise from faulty cleavage of angle structures and, as such, predispose patients to glaucoma.5,6
In isolation, posterior embryotoxon, a thickened and anteriorly displaced Schwalbe’s line visible at the corneal periphery, common and mostly benign. It can, however, be seen in conjunction with Axenfeld-Reiger syndrome in a spectrum of disorders including peripheral anterior synechiae and iris defects. Posterior embryotoxon can have associated dental, facial and skeletal abnormalities. Patients with Peter’s anomaly, a rare and serious bilateral condition, can present with features of both posterior embryotoxon and Axenfeld-Reiger, as well as central corneal opacification and iridocorneal and iridolenticular adhesions. Patients with any of the aforementioned conditions should be monitored closely for glaucoma.5-7
Edema
See discussion of congenital hereditary endothelial dystrophy (CHED) below.
Dermoids and Dystrophies
Limbal dermoids, one of three types of conjunctival choristomas, are mostly comprised of collagen and are typically located inferior-temporally at the limbus. Under slit lamp examination, a dermoid is well circumscribed, elevated and can appear fleshy to pale yellow in color. Treatment calls for excision, as the growth is superficial.3 Although limbal dermoids are present unilaterally, a bilateral presentation is typical of children with Goldenhar’s syndrome, who will also present with colobomas of the lid and iris or total aniridia.3
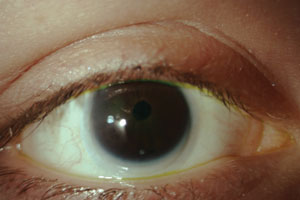 |
| Small corneal diameter in a patient with microcornea. Photo: Renee Reeder, OD |
Another strategy for diagnosis is location: dystrophies tend to target the central portion of a single corneal layer, dramatically narrowing the list of differentials from innumerable to several. Within a given layer, dystrophies can often be differentiated based on clinical presentation along with any accompanying signs or symptoms; ability to describe these will, of course, be dependent on patient age. In addition, optical coherence tomography can be a useful tool in localizing corneal changes and deposits and further aiding in diagnosis.
Generally, there are few other associated ocular or systemic findings to complicate the picture with corneal dystrophies. Here is a summary of the most common dystrophies affecting the pediatric population:4-7
Epithelial. Meesmann and Lisch epithelial dystrophies are similar entities presenting with diffuse (Meesmann) or localized (Lisch) epithelial microcysts and vesicles. As these rupture onto the epithelial surface, minor pain and opacification may be noted. The conditions are relatively benign and management is often palliative. In contrast, gelatinous drop-like dystrophy presents with pronounced central raised mounds of amyloid in the epithelium, resembling myriad fruit from mulberry (early childhood) to a late-stage yellow kumquat appearance. This condition is more likely to warrant keratectomy referral.4-7
Bowman’s layer. The three conditions classified under Bowman’s layer dystrophy were once thought to be the same etiology and only recently have been distinguished, based on microscopic appearance and clinical presentation. Reis-Bücklers, Thiel-Behnke and Grayson-Wilbrandt are autosomal dominant conditions that present in the first decade of life with bilateral, symmetric and subepithelial opacities in a honeycomb pattern. With variations in severity, treatment plans range from lubrication to keratectomy referral.4-7
Stroma. Congenital stromal dystrophy—a cousin of CHED—is a rare condition causing congenital opaque, flaky clouding of the corneal stroma. The condition often results in marked corneal edema.4-7 Granular dystrophy and lattice dystrophy are common autosomal dominant conditions leading to stromal deposition of hyaline and amyloid deposits in a diffuse and branching pattern, respectively. A third condition, Avellino dystrophy, combines features of both and is named after the original group of affected individuals from Avellino, Italy.4-7 Central cloudy dystrophy of Francois and Schnyder’s dystrophy both involve diffuse central deposition of cholesterol in the corneal stroma early in life, and the latter should be worked up for elevated serum cholesterol levels.4-7
Endothelium. Similar to congenital stromal dystrophy, CHED is also characterized by congenital bilateral corneal clouding—from mild haze to a milky appearance—with an accompanying thickened Descemet’s membrane and generalized corneal edema. This is another condition that should be differentiated from congenital glaucoma, as these patients often have high IOP readings secondary to endothelial dysfunction and thickened corneas.4-7 Posterior polymorphous dystrophy is also relatively common in the pediatric population. In this condition, endothelial cells act like epithelial cells, propagating in linear, band-like or grouped configurations. These cells invade the angle and the anterior iris, causing elevated pressure, iris distortion and abnormalities.4-7
While corneal anomalies in a pediatric population may seem daunting, an understanding of the history of the findings, as well as a clinical examination to determine the layer in which the condition is found, can significantly narrow the list of differentials. While many conditions are slowly progressive and largely benign, keeping a close eye on sight-threatening conditions, both direct (elevations in IOP) and indirect (form-deprivation amblyopia, especially in unilateral conditions), is critical in preventing long-term complications.
Dr. Fromstein completed her Doctor of Optometry degree at Nova Southeastern University in Davie, Fla., and pursued a residency in Cornea and Contact Lens at the Illinois College of Optometry, where she is currently an assistant professor and coordinator of the Cornea and Contact Lens residency.
Dr. Trujillo is an assistant professor at the Pennsylvania College of Optometry, Salus University. He specializes in pediatric care, binocular vision, strabismus and amblyopia.
1. Raab EL, Aaby AA, Bloom JN, et al. Diseases of the cornea and anterior segment. In: Skuta GL, Cantor LB, Weiss JS (eds.) Basic and clinical science course. San Francisco: American Academy of Ophthalmology; 2012:211–21.
2. Nischal KK, FRCOphth. Genetics of congenital corneal opacification-impact on diagnosis and treatment. 34 (Supplement 10):S24-S34, October 2015.
3. Yanoff M, Duker JS. Ophthalmology 2nd edition. St. Louis: Mosby; 2004.
4. Weisenthal R, Afshari N, Bouchard C, et al. Basic and clinical science course. San Francisco: American Academy of Ophthalmology: 2014:492-530.
5. Kanski JJ, Bowling B, Nischal KK, Pearson A. Clinical ophthalmology: a systematic approach. Edinburgh: Elsevier/Saunders. 2011.
6. Kaiser PK. The Massachusetts Eye and Ear Infirmary illustrated manual of ophthalmology. Philadelphia: Saunders: 2014.
7. Rapuano C. Cornea color atlas & synopsis of clinical ophthalmology. Philadelphia: Lippincott Williams & Wilkins: 2012.
8. Stretton S, Gopinathan U, Wilcox MD. Corneal ulceration in pediatric patients: a brief overview of progress in topical treatment. Paediatr Drugs. 2002;4(2):95-110.
9. Wasielica-Poslednik J, Politino G, Schmidtmann I, et al. Influence of corneal opacity on intraocular pressure assessment in patients with lysosomal storage diseases. PLoS One. 2017;12(1):e0168698.

